Introduction
Chronic granulomatous disease (CGD) is an inborn error of immunity (IEI) impairing phagocyte function caused by genetic defects in the nicotinamide adenine dinucleotide phosphate (NADPH) system of leukocytes composed of gp91phox, p22phox, p40phox, p47phox, and p67phox subunits (1). CYBB gene (encoding gp91phox) mutation is affected in X-linked; NCF-1 (p47phox), CYBA (p22phox), NCF4 (p40phox), and NCF2 (p67phox) are affected in autosomal recessive (AR form). A recently recognized CYBC1 and RAC2 gene mutations also cause CGD (2, 3). In the Western world, approximately two-thirds of CGD is inherited as an X trait, but in Turkey, most of the patients have an AR mutation due to consanguineous marriage (2, 4).
CGD patients are prone to recurrent, life-threatening infections caused by catalase-positive bacteria (i.e., Staphylococcus aureus, Burkholderia cepacia complex, Nocardia spp., and Serratia marcescens, and Aspergillus spp.) (5).
They can be diagnosed at any age, mostly under five (6). Nevertheless, they can be diagnosed in adulthood due to the incomplete penetrance and variable expressivity of IEIs, advances and increased availability in diagnostic methods, and new potent antibiotics. The identification of monogenic defects underlying IEI has increased with globally available Next-generation sequencing (NGS) tests (7). CGD incidence is approximately 1:250,000 live births (3).
We aimed to present a CGD case diagnosed at an older age with invasive pulmonary aspergillosis plus Burkholderia pneumonia and developed hemophagocytic lymphohistiocytosis (HLH) during follow-up, all successfully managed with a multidisciplinary approach.
Case
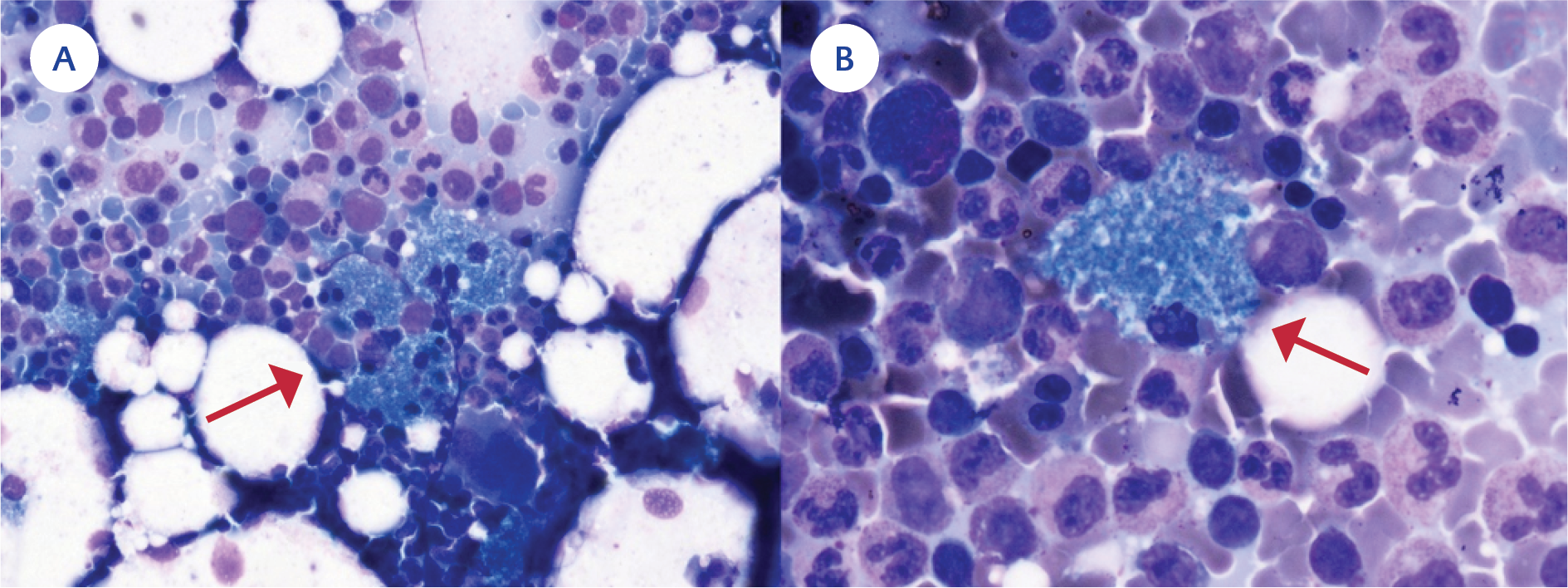
Figure 1. A) Red arrow: Bone marrow smear shows a cluster of sea-blue histiocytes (May–Grünwald–Giemsa staining ×40), B) Red arrow: Sea-blue histiocyte (May–Grünwald–Giemsa staining ×100).
A 36-year-old male with a history of type 1 diabetes mellitus (T1DM) and recurrent skin infections applied elsewhere with fever, productive cough, and dyspnea. Chest computed tomography (CT) revealed consolidation with air bronchograms; nevertheless, the pneumonia failed to respond to the moxifloxacin therapy. He was referred to our center due to persistent symptoms. As his leukocyte (31,090 /mL), procalcitonin (4.1 ng/mL), and C-reactive protein (CRP) (224 mg/L) levels indicated bacterial pneumonia, we started meropenem empirically. The respiratory multiplex polymerase chain reaction (PCR) was negative. Blood cultures remained sterile; however, bronchoalveolar lavage yielded Burkholderia multivorans and sputum culture Aspergillus niger. A follow-up chest CT demonstrated developing nodules consistent with a fungal infection (Figure 1).
Treatment was switched to voriconazole plus ceftazidime. Despite five days of treatment, the patient remained feverish, and his condition worsened. He was consulted with pediatric immunology. His family history included second-degree relative parents, the death of a 31-year-old mother due to sepsis, and frequent skin infections in his father and two siblings. His sister was also diagnosed with CGD later on.
His peripheral blood lymphocyte subsets and immunoglobulin levels were normal. There was no oxidative activity in the dihydroergotamine-123 (DHR) test. Nitroblue tetrazolium (NBT) test result was 0. Hence, a CGD diagnosis was made while waiting for the genetic test. Interferon-gamma (IFN-γ) (50 mcg/m2, thrice weekly) was started. Meanwhile, follow-up blood cultures yielded B. multivorans/cepacia complex, and he developed hepatosplenomegaly (liver size 17 cm, spleen size 15 cm), deepening anemia (hemoglobin from 11.5 to 9 g/dL), increased CRP (415 mg/L), hyperferritinemia (1170 mcg/L), and elevated triglycerides (1173 mg/dL).
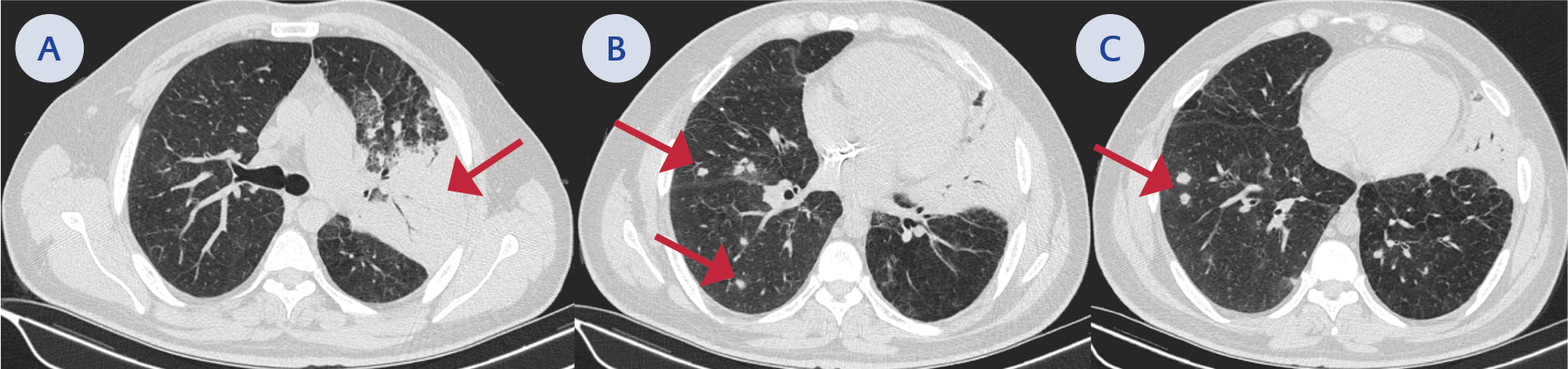
Figure 2. A) Red arrow: consolidation with air bronchograms in the left upper lobe, B-C) Developing nodules consistent with a fungal infection.
Thus, HLH due to B. cepacia infection was considered; intravenous immunoglobulin (30 g), methylprednisolone (60 mg/dL), and trimethoprim-sulfamethoxazole (thrice a day, 5 mg/kg trimethoprim) were initiated. The following day, his fever went down, his general condition improved, and marker levels declined dramatically (ferritin: 470 mcg/L, triglyceride 611 mg/dL, CRP 101 mg/L). A bone marrow aspiration and biopsy showed sea blue histiocytes favoring CGD (8) (Figure 2). A thyroid ultrasound revealed thyroiditis. Whole exome sequencing confirmed a GT deletion in the GTGT repeat in exon 2 of the NCF1 gene. It was confirmed by the Sanger sequencing method (Figure 3) (9,10).

Figure 3. Schematic representation of NCF1 protein showing its domains and localization of the mutation identified in the patient. GT deletion (c.75_76delGT) in the GTGT repeat in exon 2 of the NCF1 gene is very common and is caused by a crossover between the NCF1 and one of its two pseudogenes.
He was discharged with subcutaneous IFN-γ, voriconazole, trimethoprim-sulfamethoxazole, and methylprednisolone on a tapering regimen. Allogeneic hematopoietic stem cell transplantation (aHSCT) was planned.
Discussion
CGD is characterized by immunodeficiency, dysregulated inflammation, and increased autoimmunity (1). An NCF1 deletion was detected in our patient, which was compatible with AR penetration. Although X-linked inheritance is observed in 65-75% of cases, the AR pattern frequency increases in regions with common consanguineous marriages (11). AR form presents at a later age, with a milder clinical presentation (2, 4).
Formerly, it was believed that IEIs followed a Mendelian pattern with complete penetrance. Expanded NGS usage improved our understanding of disease penetrance and expressivity. Thus, CGD cases with milder presentation began to be diagnosed at older ages (7). Barkai et al. reported 16 patients with CGD diagnosis 19 years and above (median age was 38.5 [19-74]) (12). Our patient was diagnosed at 36, and his sister at 40.
CGD’s most common infection sites are the lungs, liver, lymph nodes, bones, and skin (5). Our patient had recurrent skin infections and pneumonia. HLH is a condition of excessive immune activation and can be seen in CGD due to persistent infection or dysregulated immune response (13). He had possible HLH. Even if he met 4 of 8 HLH-2004 criteria, he was treated since HLH can be mortal and should be treated immediately.
CGD patients have a higher risk of autoimmune disorders. NAPDH oxidase-derived reactive oxygen species may have immunoregulatory effects, and their deficiency may cause autoimmunity (2, 14). Our patient had T1DM and thyroiditis.
CGD treatment involves prophylaxis with trimethoprim-sulfamethoxazole, itraconazole, and IFN-γ. IFN-γ was shown to reduce the number and severity of infections in CGD patients for all genetic types (15). However, its effect on autosomal recessive forms is still debatable (16), so more clinical trials are needed to evaluate the efficacy of IFN-γ. Although these treatments have significantly improved CGD prognosis recently, aHSCT still remains the definitive treatment (2).
Conclusion
Screening with NBT and DHR tests may help diagnose CGD in patients with a history of frequent infections, Aspergillus spp. growth in any culture without a known immunosuppressive disease, and Burkholderia spp. growth in the absence of cystic fibrosis. This case report contributes to understanding CGD in adults, aiming to enhance diagnostic and therapeutic strategies.